
- Academic Editor
The genome of the Nipah virus (NiV) encodes a variety of structural proteins linked to a diverse array of symptoms, including fevers, headaches, somnolence, and respiratory impairment. In instances of heightened severity, it can also invade the central nervous system (CNS), resulting in more pronounced problems. This work investigates the effects of NiV on the blood-brain barrier (BBB), the vital physiological layer responsible for safeguarding the CNS by regulating the passage of chemicals into the brain selectively. To achieve this, the researchers (MMJAO, AM and MNMD) searched a variety of databases for relevant articles on NiV and BBB disruption, looking for evidence of work on inflammation, immune response (cytokines and chemokines), tight junctions (TJs), and basement membranes related to NiV and BBB. Based on these works, it appears that the affinity of NiV for various receptors, including Ephrin-B2 and Ephrin-B3, has seen many NiV infections begin in the respiratory epithelium, resulting in the development of acute respiratory distress syndrome. The virus then gains entry into the circulatory system, offering it the potential to invade brain endothelial cells (ECs). NiV also has the ability to infect the leukocytes and the olfactory pathway, offering it a “Trojan horse” strategy. When NiV causes encephalitis, the CNS generates a strong inflammatory response, which makes the blood vessels more permeable. Chemokines and cytokines all have a substantial influence on BBB disruption, and NiV also has the ability to affect TJs, leading to disturbances in the structural integrity of the BBB. The pathogen’s versatility is also shown by its capacity to impact multiple organ systems, despite particular emphasis on the CNS. It is of the utmost importance to comprehend the mechanisms by which NiV impacts the integrity of the BBB, as such comprehension has the potential to inform treatment approaches for NiV and other developing viral diseases. Nevertheless, the complicated pathophysiology and molecular pathways implicated in this phenomenon have offered several difficult challenges to researchers to date, underscoring the need for sustained scientific investigation and collaboration in the ongoing battle against this powerful virus.
The Nipah virus (NiV) lies within the Paramyxoviridae family, part of the Henipavirus genus. It is characterised as a single-stranded RNA virus with a negative-sense genome [1] that encodes six distinct structural proteins (Fig. 1): nucleoprotein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), attachment glycoprotein (G), and the large protein or RNA polymerase protein (L). Infections of the central nervous system (CNS), cardiovascular system, kidneys, and lungs have all been linked to NiV [2], which is thus associated with a wide range of side effects; however, the most well-known ones include fever, headache, and sleepiness [3]. Initial infections have also been linked to breathing difficulties [3].
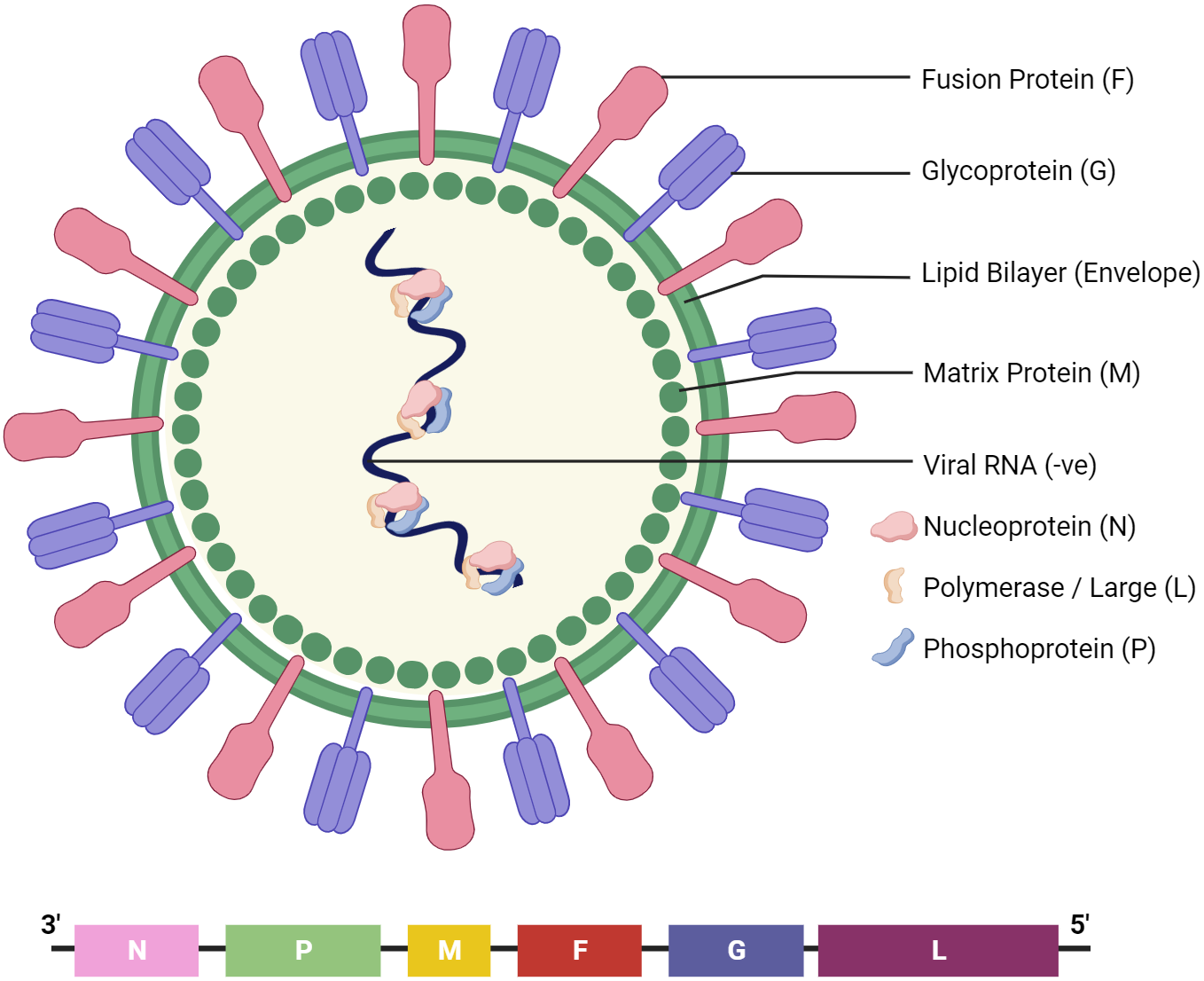 Fig. 1.
Fig. 1.The structure of Nipah virus (NiV). It encodes six structural proteins: nucleoprotein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), receptor-binding glycoprotein (G), and RNA polymerase protein (L).
The blood-brain barrier (BBB) is a physiological layer that effectively prevents the unregulated leakage of endogenous substances and xenobiotics into the brain, thereby preserving the homeostatic neural microenvironment crucial for the proper functioning of neurons. The inner lining of brain microvessels consists of microvascular endothelial cells (ECs), which form the BBB. These cells lie in close proximity to the basal membrane, surrounded by astrocytic end-foot processes and accompanied by pericytes. The BBB has the characteristic of selective permeability, which enables it to permit the passage of particular substances while impeding others, hence safeguarding the integrity of the CNS. It thus hinders the entry of various molecules, safeguarding the brain’s parenchyma from infection while facilitating the outflow of waste molecules. The BBB thus serves the purpose of maintaining the optimal fluid environment required for proper brain functioning alongside safeguarding the CNS tissue [4].
While the mechanism by which NiV enters the CNS is not presently known, ECs are a prominent target of this virus [3]. Ephrin-B2 and ephrin-B3 are viral entrance receptors that are expressed on a wide variety of cell types in various organs [5]. Prior studies have demonstrated in vivo expression of Ephrin-B2 on ECs [6], while neurons and brain endothelium both express the tyrosine kinase receptors Ephrin-B2 and Ephrin-B3 [7, 8]. The ligand Ephrin-B2 has further been identified as an anchor for EphB4 receptors, and this is involved in the processes of angiogenesis and neurogenesis [9]. Other work provides evidence of ephrin-B3 expression inside the brain parenchyma, indicating its potential role as an alternative receptor in certain cell types when Ephrin-B2 is inaccessible [10]. Additionally, the virus may gain entry into specific cells by micropinocytosis [11]. Macropinocytosis is a cellular mechanism of endocytosis characterised by the random internalisation of substantial quantities of extracellular fluid, solutes, and membrane into enlarged endocytic vesicles known as macropinosomes [12]. After this process, the virus releases its genetic material into the cytoplasm of the cell by undergoing conformational alterations in the G and F proteins.
While NiV’s route and speed of entry into the CNS and its subsequent spread throughout the host are poorly understood, most studies appear to agree that several mechanisms may occur that facilitate NiV entry into the CNS by allowing infected cells to cross the BBB [2]. One theory is that the BBB can be disrupted by a Trojan horse method that directly infects lymphocytes and monocytes with the NiV virus [2]. Previous research has also revealed that NiV has the ability to infect neurons and microglial cells that exhibit CD68 in nonhuman primates [13].
The purpose of the current review was thus to investigate the process of NiV infection and the effects of this virus on the BBB and its degradation as discussed in the literature. Immune response leading to BBB breakdown and cellular mechanism by which NiV triggers BBB breakdown was also investigated, alongside the integrity of the tight junctions (TJs), basement membrane, and adhesion molecules, all of which may be impaired by NiV infection, causing BBB breakdown. This was undertaken as the development of effective techniques for the management and control of NiV infection may benefit from a better understanding of such virus and host interactions.
For this work, three researchers, MMJAO, AM and MNMD, conducted a literature search using the Google Scholar, PubMed, and EMBASE databases in order to identify papers relevant to the study. The search was conducted for the period from January 2018 to June 2023 using specific keywords in order to identify relevant studies recently published in the English language. The keywords utilised to achieve this included variations such as “BBB”, “Blood brain barrier”, “Brain”, and “BBB degradation”. The search focused, however, on topics related to “Nipah virus” and “Paramyxoviridae and Henipavirus”, with additional filters for “immune response”, “inflammation markers”, “cytokines”, and “chemokines”. A further search was then added for “tight junctions and adhesion molecules” along with “endothelial barrier”. The study focused on the relationships between inflammation, cytokine storm, pro-inflammatory cytokines, immune response, and TJs, in conjunction with the effects on the basement membrane related to BBB disruption by NiV.
In order to identify supplementary papers aligned with the research requirements that were not initially retrieved, the reference lists of publications arising from the original search were additionally examined. The study employed the following inclusion criteria: (1) articles written in English; (2) research focused on the NiV and the BBB; (3) observational studies conducted retrospectively or prospectively; (4) studies investigating acute therapy for the NiV; and (5) assessments of BBB disruption conducted prior to the administration of acute NiV treatment. The study thus included works with both experimental and animal research methodologies, as well as data from the World Health Organization (WHO) and Centers for Disease Control and Prevention (CDC) websites. Where there was overlap across study cohorts, only the research with the largest sample size was considered. Conference papers, research procedures, and unpublished works were not considered for inclusion.
NiV is a member of the Paramyxovirus family of negative-sense, single-stranded RNA viruses. Paramyxoviruses are generally thought of as viruses with a limited host range and a propensity for causing only mild illness outbreaks [14]. NiV is most usually spread by fruit bats (Pteropus species), though there has been a rise in human-to-human transmission in recent years [15]. In contrast to the usual mild Paramyxovirus severity, NiV epidemics cause severe illnesses with significant fatality rates in the southeast Asian area almost every year. Human and pig infections with NiV were first documented in 1998 in Malaysia, where the spread of the virus to wider parts of the country was attributed to the movement of the sick pigs. During 1998, NiV spread from pigs to humans, and only after the slaughter of 900,000 suspected infected pigs was that the outbreak contained [16]. Nipah thus became the name commonly given to the virus, after the Malaysian village where it was initially discovered [17]. In subsequent occurrences in Bangladesh and India, the transmission of NiV was believed to be via direct human-to-human contact, primarily via individuals’ secretions and excretions [15, 18]. In the city of Siliguri, India, in 2001, instances of viral transmission were documented inside a healthcare facility, with over 75% of the reported cases in hat outbreak seen among individuals affiliated with the hospital, including staff members and tourists. Australia, Bangladesh, Cambodia, China, India, Indonesia, Madagascar, Taiwan (China), and Thailand have all had outbreaks, based on serological evidence or genetic detection in pteropus bats, and Bhutan, Brunei, China, Indonesia, Laos, Madagascar, Myanmar, Nepal, the Philippines, Singapore, Taiwan (China), Thailand, and Vietnam are all home to endemic populations of pteropus bats [19].
The duration of clinical symptoms in patients infected with NiV is variable, with the severity of the disease encompassing the spectrum from asymptomatic (subclinical) infection to acute respiratory illness and encephalitis. The onset of NiV disease is typically marked by influenza-like symptoms, including fever, muscle pain, cough, vomiting, and headaches [20]. Individuals may experience respiratory difficulties characterised by symptoms such as a feeling cold, respiratory distress, shortness of breath, and atypical pneumonia. NiV infection causes also localised alterations in blood vessels, resulting in the development of vasculitis in the tiny vessels in humans. An unfavourable prognosis is associated with advanced age, thrombocytopenia, or the coexistence of additional comorbidities [21]. In humans, infection primarily spreads through the mouth and nose, with various incubation durations documented in the literature, commonly ranging from one to two weeks, though potentially anywhere from four to 21 days [22]. Typically, symptoms are not apparent until after a clinically inactive period of four days to two weeks [23].
Furthermore, as the virus disseminates to vital organs [24] during the severe phase, NiV-infection has the potential to infiltrate the CNS, resulting in varying levels of encephalopathy. The brain tissue then commonly exhibits necrotic plaques and occasionally syncytia composed of multinucleated large ECs. Neurological symptoms including seizures, impaired cerebellar function, and diminished consciousness may then ensue [25]. Instances of cognitive confusion have also been observed [3]. After a week, the typical symptoms of encephalitis may include decreased muscle tone (hypotonia), involuntary muscle contractions in specific areas (segmental myoclonus), absence of reflexes (areflexia), weakness in the limbs, and inability to control eye movements (gaze palsy). Subsequently, coma and mortality may ensue within a matter of days, and the fatality rate of NiV infection is severe, with approximately 40 to 75% of infected individuals [26], however, with reported death rates during NiV epidemics reaching as high as 95% [27]. Approximately 20% of those individuals who recover from acute infection may also experience persistent fatigue, neurological impairments, and depression [28], and there have been reported cases of recurrent or late-onset encephalitis [29].
Meanwhile, the NiV has the ability to endure in the body for a considerable duration, since it has been shown to survive and persist in the bloodstream for a maximum of 7 days [30]. The virus has demonstrated the ability to remain alive in tissue culture conditions for a minimum duration of 14 days. Additional investigation is required to have a more comprehensive understanding of the mechanisms behind the viral persistence. Autopsy studies on humans have revealed that acute NiV infection is a widespread illness that affects blood vessels and cells in most major organs, including the brain [31]. Various risk variables were found to have a substantial association with NiV infection, such as engaging in tree climbing, being male, traveling outside of one’s own sub-district, and being exposed to date palm sap or pigs [27].
When diagnosing this particular NiV, a number of different practical procedures are utilized. The competitive enzyme-linked immunosorbent assay (cELISA) is the serological test used to diagnose NiV infection [32]. This uses a monoclonal antibody and recombinant Nipah glycoprotein as antigens, achieving high diagnostic sensitivity (98.58%) and specificity (99.92%), with the ability to detect antibodies from 14 days after infection. Another frequently employed serological assay is the plaque reduction neutralization test (PRNT). This test remains positive throughout the whole trial and demonstrates 100% consistency with the desired outcome. Additionally, real-time polymerase chain reaction (PCR) is another common method used for identifying pathogens by means of specific primers and probes. Furthermore, ELISA is also used to identify antibodies against NiV in patient samples, based on colorimetric or fluorescence signals. Moreover, immunofluorescence assays use fluorescently tagged antibodies to identify viral antigens, while histopathology can detect alterations linked to NiV infection, such as inflammation and cellular injury. Virus isolation and neutralisation can be done using cell culture techniques. Lastly, a new biosensor, (DNAzyme-Based Fluorescent Biosensor) NiV, has also been developed to detect NiV RNA, based on a split catalytic core of deoxyribozyme 10-23. This offers rapid and precise identification, with a detection threshold of 10 nM for synthesized target RNA [33].
As noted, the BBB plays a crucial role in regulating the transportation of solutes, chemicals, and cells, with the primary objective being to limit the infiltration of pathogens into the brain. The BBB is a dynamic entity that may undergo modulation through the influence of signalling molecules. However, one hypothesis suggests that some viruses have the potential to infiltrate the CNS via hematogenous, neural, retrograde axonal transport, and the olfactory nerve, as various chemicals synthesised by the endothelium, astrocytes, and neurons in proximity to the capillaries have been discovered to modulate the permeability of the BBB. Cytokines, chemokines, and neurotransmitters are all examples of BBB-permeable chemicals [34], and these are significant due to the fact that there is some evidence that systemic inflammation can cause a slight rise in BBB permeability, putting the CNS at a higher risk of infection [35, 36]. In addition, it is widely recognised that TJs play a crucial role in the formation and maintenance of the BBB, as well as in regulating the movement of viruses through the paracellular route. TJs, which are established by the interaction of adjacent ECs, thus play a significant role in the establishment of a physical barrier [37, 38]. Pericytes therefore play a crucial role in preserving the structural and functional integrity of the BBB by supporting the TJs of ECs, such as claudin-5, occludin, and Zonula occludens-1 (ZO-1), and by regulating the process of transcytosis in ECs [37, 38]. Paradis et al. [39] also reported that several bacterial and viral infections have the potential to negatively impact TJs through various mechanisms, resulting in BBB disruption.
The transmission dynamics of NiV inside an infected human host have been subject to only minimal investigation, thereby necessitating emphasis on insights derived from animal models. In hamsters, for instance, NiV was discovered to infect the olfactory epithelium of the respiratory system first [40] before entering the brain via the olfactory pathway, following the neurons of the olfactory epithelium [41]. Additionally, the cranial nerves and the bloodstream both appear to play a role in NiV’s dissemination into the CNS in the pig model [42]. Recent in vivo research confirmed that NiV replicated successfully in human olfactory ECs and sensory neurons, suggesting that this approach may also be possible for human infection [43]. However, NiV may instead infect and disseminate itself via leukocytes, as NiV replication at low levels has been shown in both a monocytic cell line (THP-1) and immature dendritic cells [44, 45]. According to reports by Mathieu et al. [45], NiV may attach to the cell surface of lymphocytes and monocytes, leading to trans-infection in vitro in hamsters. It is also possible for infected dendritic cells to function as a “Trojan horse” and successfully traverse the BBB, as shown by Tiong et al. [2].
Invasion of the host cell, viral assembly, budding, and immune evasion are all factors that contribute to a virus’s pathogenicity, yet overall understanding of the NiV pathogenesis remains elusive. The host cell receptors, ephrin-B2 and ephrin-B3, have been identified as the causative agents for infection by the NiV virus in both animal and human hosts [46]. The entrance of NiV is facilitated by a pH-independent process of membrane fusion, which is mediated by the attachment and fusing of glycoproteins. In addition, the ligands ephrin-B2 and ephrin-B3, which belong to the family of receptor tyrosine kinases, have been observed to play a role in cell fusion and thus to determine the host cell’s susceptibility to NiV [46]. NiV-binding sites are modulators of cell migration in the nervous system, located on the surface of most mammalian ECs, though the highest levels of expression are in the lungs. These receptors are widespread among mammals, contributing to the spread of the disease: when infectious virus particles are breathed in, infection begins.
ECs have been shown to be the primary target cells following inhalation, though CNS infection has also been documented. However, full comprehension of the process remains elusive. Acute respiratory distress syndrome is a consequence of NiV infection of the respiratory epithelium, which triggers the release of inflammatory cytokines and the subsequent recruitment of immune cells. The Interlukine (IL)-6, IL-8, and IL-1, as well as Monocyte Chemoattractant Protein-1 (MCP-1), granulocyte colony-stimulating factor, and granulocyte-monocyte colony-stimulating factor, are all important mediators [47]. Replication and dissemination of the virus occur inside the respiratory epithelium, followed by transfer to the endothelium. NiV may bind to CD3 leukocytes [46], as well as infecting monocytes, CD6, CD8 T lymphocytes, and natural killer cells, as seen in a pig investigation [46].
The microvascular ECs that form the BBB contain active leukocyte adhesion
molecules to which CD6 binds. These adhesion molecules are involved in the
regulation of immune responses in the CNS and are essential for the diapedesis of
immune cells across the BBB [48]. Key adhesion molecules present on BBB-ECs
include Activated leukocyte cell adhesion molecules, Melanoma cell adhesion
molecules, Mucosal vascular addressin cell adhesion molecule 1, Vascular adhesion
protein (VAP-1), Junctional adhesion Molecule-L, and Ninjurin-1 [48]. The extent
to which the NiV gains access to the CNS following its interaction with CD6 is
uncertain. Once the BBB is breached, the NiV can infiltrate the brain and induce
encephalitis by various routes, including the olfactory nerve, the choroid
plexus, or the cerebral arteries. It is worth noting that the permeability of the
BBB has been associated with inflammatory cytokines including tumour necrosis
factor-alpha (TNF-
NiV pathophysiology and its molecular basis remain poorly understood, though the immune response is identified by the synthesis of pro-inflammatory cytokines, chemokines, and interferons, which aid in the elimination of the virus, but which may also result in tissue damage [52]. NiV can also trigger apoptosis in the cells it infects, exacerbating tissue damage and the advancement of the disease [53]. In order to control NiV infection and stop viral spread to other organs, the adaptive immune response is triggered by activating T cells and producing antibodies that are specific to the virus [54]. Nevertheless, immunopathology, which can result from dysregulated immune responses, appears to amplify the impact of NiV-induced encephalitis [54]. Acute NiV encephalitis is further characterised by neuroglial infection and ischaemia-microinfarction associated with vasculitis-induced thrombotic occlusion [55]. Human NiV infections are further characterised by widespread infection of neurons, ECs, and smooth muscle cells in the arteries [20]. Humans infected with NiV develop a severe form of encephalitis marked by systemic vasculitis and necrosis, particularly in the CNS [56].
Again, during NiV encephalitis, the CNS is flooded with inflammatory cells such
as neutrophils, macrophages, lymphocytes, and reactive microglia [20]. It is
conceivable that cytokines and chemokines, which act as small molecule messengers
and chemoattractants, are responsible for the attraction of these cells. The
chemokines IL-8, C-X-C motif chemokine ligand-8 (CXCL8), regulated upon
activation, normal T cell expressed and secreted (RANTES) also know as CCL5,
macrophage inflammatory protein-1 (MIP-1) also known as CCL3, MCP-1 also known as CCL2,
and C-X-C motif chemokine ligand-10 (CXCL10), also known as interferon
gamma-induced protein-10 (IP-10), as well as the inflammatory cytokines TNF and
IL-6, have all been observed in viral CNS disease [54, 57, 58]. Cells in the CNS
and the inflammatory infiltrate may also both manufacture and release cytokines
[59, 60], highlighting the diversity of cellular sources for these molecules.
Chemokines in particular play a crucial role in directing cell migration.
Previous data further suggests that NiV infection of human lung and lung
lymphatic ECs leads to increased expression of the inflammatory chemokines MCP-1,
IL-8, and IP-10, which may play a role in the activation of monocyte and T
lymphocyte chemotaxis [56]. Additionally, it is demonstrated that smaller airway
epithelium becomes infected, it triggers the production of important inflammatory
mediators such as interleukin IL-1
Two mechanisms illustrate the direct actions of cytokines crossing the BBB in
quantities that cause CNS effects [63]. The first is Transporter-mediated
transcytosis, in which transporters that are specifically expressed on the ECs of
the BBB aid in the transportation of cytokines across the barrier. These
transporters have the ability to identify and attach to cytokines, facilitating
their movement from the bloodstream to the brain side of the BBB. This mechanism
is reliant on energy and necessitates the participation of vesicular pathways.
The transporters implicated in this method have the ability to control the
expression, location, and conformation of the transport process. Among these,
solute carriers (SLCs) are a group of transporters that have a vital function in
transporting different compounds, such as cytokines, through the BBB. The second
mechanism, Receptor-mediated transcytosis, is a process that allows chemicals
such as cytokines to traverse the BBB by binding to specific receptors on the ECs
of the BBB. This mechanism entails the attachment of cytokines to their
corresponding receptors on the luminal (blood-facing) side of the BBB’s ECs.
After such binding, the cytokine-receptor complex is taken up into vesicles
through endocytosis, and the vesicles thereafter undergo intracellular
trafficking, potentially traversing organelles such as endosomes or the Golgi
apparatus. Ultimately, the vesicles that hold the cytokine-receptor complex are
conveyed via the ECs and discharged on the abluminal side of the BBB, enabling
the cytokines to penetrate the CNS. Cannabinoid type 2 receptors (CB2 receptors)
are present in brain endothelial cells (BECs), and these are involved in
regulating the BBB response to neuroimmune stimuli. Stimulation of CB2 receptors
can thus hinder the increase in gene expression caused by TNF-
Meanwhile, previous studies have shown that primary human ECs infected with NiV
can generate a functional interferon-beta (IFN-
Furthermore, signal transducer and activator of transcription 1 (STAT1) and STAT2 are proteins which play crucial roles in the the
Janus kinase/signal transducers and activators of transcription (JAK-STAT)
signalling pathway, a pivotal signalling cascade involved in various cellular
processes, including immunological responses and cellular development regulation.
The collective action of P, V, and W proteins results in the inhibition of
STAT1/STAT2, thereby affecting the signal transduction of IFN-
The initiation of primary brain microvascular ECs activation can occur at an
early stage, triggered by the stimulation of virus-like particles carrying NiV
glycoproteins, regardless of viral replication [69]. Previous studies have
demonstrated that the process of binding and fusing of particles carrying the NiV
glycoproteins leads to the rapid induction of upregulation in adhesion molecules,
particularly E-selectin [69]. E-selectin is involved in the process of leukocyte
recruitment to the endothelium in the context of inflammation [70]. The
upregulation of E-selectin thus occurs prior to the production of various
cytokines, including IFN-
Moreover, TJs also play a crucial role in preserving the structural integrity of polarised epithelial tissues. Certain viruses have the ability to modify the barrier function of the epithelia they invade, enabling them to traverse the barrier and thus to spread throughout the host organism. This necessitates the alteration or deterioration of TJ [73], and the TJ of polarized Caco-2 cells infected with rotavirus exhibit changes, characterised by the disorganisation of occludin [74]. Enveloped viruses possess fusogenic properties due to the presence of glycoproteins and fusion proteins, and the fusion of cells infected with NiV results in the disruption of TJ [75]. Lamp et al. [75] demonstrated that the M protein is responsible for facilitating the process of viral assembly and release through the apical membrane: while NiV F and G proteins exhibit initial localisation to the basolateral membrane and elicit cellular fusion, the M protein is synthesised inside the cytoplasm of the host cell. Following cell union, there is thus a subsequent apical buildup of M, F, and G proteins, resulting in the eventual apical release of NiV particles. This causes the epithelial barrier’s structural integrity to be compromised, resulting in the release of NiV from the basolateral side and its subsequent diffusion throughout the host [75], increasing vascular permeability and causing the rupture of TJs, ultimately generating disruption of the BBB.
In terms of NiV encephalitis and disruption of the BBB, several potential causes of viral tropism that may subsequently lead to BBB disruption have thus been identified, as shown in Table 1 (Ref. [2, 5, 13, 20, 40, 41, 42, 43, 44, 45, 46, 55, 56, 62, 65, 66, 68, 69, 71, 75]). In addition, the pathogenesis of NiV is also depicted in Fig. 2.
 Fig. 2.
Fig. 2.It shows the pathophysiology of NiV. (1) Bronchial epithelial
cells harbour NiV during the initial stages of infection. (2) The bronchi and
alveoli contain NiV antigen. (3) When the airway epithelium becomes infected,
inflammatory mediators are released. (4) The virus makes its way to the pulmonary
endothelial cells (ECs) in due time. The brain is susceptible to viral infections
that can be transmitted through the bloodstream (5). (6 and 7) Viruses can
infiltrate the CNS by hematogenous and anterograde olfactory nerves. G-CSF,
granulocyte-colony stimulating factor; MCP-1, Monocyte chemoattractant Protein-1; IL-1
| Author | Year | Type of study | Neurological Disorder | Outcome |
| Escaffre et al. [5] | 2013 | In vivo | Encephalitis | Ephrin-B2 and ephrin-B3 are Nipah viral entrance receptors expressed on a wide variety of cell types in various organs. |
| Liu et al. [13] | 2019 | In vivo | Encephalitis | NiV has the ability to infect neurons and microglial cells that exhibit CD68 in nonhuman primates. |
| Baseler et al. [40] | 2016 | In vivo | Eespiratory and neurological disease | NiV infects the olfactory epithelium of the respiratory system first, entering the brain via the olfactory pathway, following the neurons of the olfactory epithelium. |
| Munster et al. [41] | 2012 | In vivo | Encephalitis | |
| Borisevich et al. [43] | 2017 | In vitro | Encephalitis | NiV replicates successfully in human olfactory epithelial cells and sensory neurons. |
| Weingartl et al. [42] | 2005 | In vivo | Invasion CNS by NiV | The cranial nerves and the bloodstream both play a role in NiV dissemination into the CNS in a pig model. |
| Chang et al. [44] | 2006 | In vitro | Nipah virus infection | NiV replication at low levels shown in THP-1 and immature dendritic cells. |
| Mathieu et al. [45] | 2011 | In vivo and In vitro | Severe encephalitis | |
| Wong et al. [20] | 2002 | In vivo and In vitro | Acute encephalitis | The CNS can be flooded with inflammatory cells such neutrophils, macrophages, lymphocytes, and reactive microglia upon infection with NiV. |
| Escaffre et al. [5] | 2013 | In vivo | Encephalitis | Infected smaller airway epithelium produces essential inflammatory mediators as IL-1 |
| Shaw et al. [65] | 2005 | In vitro | NiV infection | The presence of NiV W protein in the nucleus is necessary to inhibit TLR-3 signalling, thereby enhancing the production of (TRIF). |
| Tsimbalyuk et al. [66] | 2020 | Review Article | Henipavirus Infection | The collective action of P, V, and W inhibits STAT1/STAT2, thereby affecting the signal transduction of IFN- |
| Keiffer et al. [68] | 2020 | In vitro | NiV infection | NiV proteins P, V, and W interact with STAT1, STAT2, and STAT4 to prevent INF-induced tyrosine phosphorylation. |
| Lo et al. [56] | 2010 | In vitro | Encephalitis | NiV infection of human lung and lung lymphatic endothelial cells leads to increased expression of the inflammatory chemokines MCP-1, IL-8, and IP-10, which may play a role in the activation of monocyte and T lymphocyte chemotaxis. |
| Humans infected with NiV develop a severe form of encephalitis marked by systemic vasculitis and necrosis, most noticeably in the CNS. | ||||
| Sayed et al. [46] | 2019 | Narrative review | NiV properties and its epidemiological determinants | NiV infects monocytes, CD6, CD8 T lymphocytes, and natural killer cells in pig investigations. |
| NiV can bind to CD3 leukocytes. | ||||
| Ong et al. [55] | 2022 | In vivo | Encephalitis | Acute NiV encephalitis has neuroglial infection, ischaemia-microinfarction, and vasculitis-induced thrombotic occlusion. |
| Rockx et al. [62] | 2011 | In vivo | BBB disruption | NiV-induced CNS disrupts the BBB and IL-1 |
| Tiong et al. [2] | 2018 | In vitro | Acute encephalitis | BBB can be disrupted by means of a Trojan horse method where NiV directly infects lymphocytes and monocytes. |
| Freitag & Maisner [69] | 2016 | In vitro | NiV infection | Binding and fusing of particles carrying the NiV glycoproteins leads to rapid induction of upregulation in adhesion molecules, particularly E-selectin. |
| Silva et al. [71] | 2018 | In vitro | General infection, inflammation, and immunotherapy | The upregulation of E-selectin occurs prior to the production of cytokines such as IFN- |
| Lamp et al. [75] | 2013 | In vitro | NiV infection | The fusion of cells infected with the NiV results in the disruption of TJ. |
| The M protein is responsible for facilitating the processes of viral assembly and release through the apical membrane. |
THP-1, monocytic cell line; CNS, Central nervous system; MCP-1, Monocyte chemoattractant Protein-1; IL-8,
Interlukine-8; IP-10, Interferon gamma-induced protein-10; IL-1
Although there has been some progress in the understanding of NiV infections, there remain several important areas of research that still need to be addressed fully, most notably with reference to the processes by which NiV enters the CNS. The precise pathways and mechanisms by which NiV enters the CNS remain unknown, which hinders the development of specific therapeutic treatments. It is also necessary to conduct further research on the specific host cell receptors, the interactions between viral proteins, and the full range of effects on the BBB. Future research should examine the molecular mechanisms that allow NiV to enter the CNS, the roles of different cell types, and BBB interactions to develop effective treatments for NiV-related neurological issues. The key to expanding understanding of NiV pathogenesis and improving the ability to tackle this powerful viral threat thus lies in addressing these research gaps.
NiV is currently a complex and unknowable challenge in terms of human understanding of viral infections. Its capacity to affect multiple organ systems, particularly the CNS, showcases its versatility as a pathogen. As the BBB acts as a guardian of the brain, selectively allowing or denying entry to various substances, including viruses, NiV’s ability to breach this barrier has become the subject of intense research; however, the underlying mechanisms of this are yet to be fully elucidated. Several studies have demonstrated that NiV may enhance the inflammation markers that lead to BBB disruption, while other studies have shed light on certain leukocyte populations that may aid the virus in crossing the BBB, emphasising the role of host immunity in viral dissemination. This review underscores the importance of understanding the mechanisms by which NiV affects BBB integrity, highlighting the disruption of TJs and cellular adhesion as key issues in this process. As comprehension of NiV and its interaction with the BBB deepens, new potential therapeutic strategies for NiV and other viral infections may emerge, offering a brighter horizon for the fight against emerging pathogens. Nonetheless, the complex pathogenesis of NiV and the molecular basis of its impact on the CNS continue to challenge researchers, underscoring the necessity for ongoing scientific inquiry and collaboration to address the impact of this challenging virus.
Contributions to the research were made by MMJAO in the following areas: conceptualization, methodology, data acquisition, original draft writing. In addition to providing the reviewers with some more information, AM was able to illustrate the figures. MMJAO and MNMD investigated and resolved questions about the accuracy of the article; revised and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.