
- Academic Editor
†These authors contributed equally.
Background: Ovarian cancer is a highly lethal gynecologic malignancy. ARHGAP10, a member of Rho GTPase-activating proteins, is a potential tumor suppressor in ovarian cancer. However, its role and the involved mechanism need further examination. Here, we investigated whether ARHGAP10 is also associated with ferroptosis. Methods: Lentivirus infection was used for gene overexpression or silencing. Real-time polymerase chain reaction (RT-PCR) and Western blot were used to assess mRNA and protein levels, respectively. Cell viability was assessed by Cell Counting Kit-8 (CCK-8) assay. Lipid reactive oxygen species level was measured by flow cytometry. A tumorigenicity assay was performed to evaluate tumor growth in vivo, and sections of mouse tumor tissues were examined by immunofluorescence microscopy. Chromatin Immunoprecipitation (ChIP) assay was used to assess the binding of H3K9ac to the promoter region of ARHGAP10. Results: ARHGAP10 overexpression promoted ferroptosis in ovarian cancer cells, resulting in decreased cell viability, and increased lipid reactive oxygen species (ROS) level. Further, it decreased and increased GPX4 and PTGS2 expression, respectively, and also induced suppression of tumor growth in mice. Fer-1, a potent inhibitor of ferroptosis, suppressed the above effects of ARHGAP10. Contrarily, ARHGAP10 silencing alleviated ferroptosis in ovarian cancer cells, which was reversed by RSL3, a ferroptosis-inducing agent. Lastly, sodium butyrate (SB) was found to transcriptionally regulate ARHGAP10, thereby also contributing to the ferroptosis of ovarian cancer cells. Conclusions: Our results suggest that SB/ARHGAP10/GPX4 is a new signaling axis involved in inducing ferroptosis in ovarian cancer cells and suppressing tumor growth, which has potential clinical significance.
Ovarian cancer is the second most common and fatal gynecologic malignancy [1]. There is a paucity of effective screening methods for ovarian cancer. Owing to the nonspecific symptoms in the initial stages, most cases of ovarian cancer are diagnosed at an advanced stage [2]. Surgery, platinum-based chemotherapy and targeted therapy are common treatment options for ovarian cancer. However, the mortality rate due to ovarian cancer has not significantly declined over the last four decades. The occurrence of chemoresistance also contributes to the increased mortality of patients with ovarian cancer. Indeed, ovarian cancer cells have an increased antioxidant capacity and express membrane transporters that can pump out the drug from the cells [3]. Therefore, in-depth characterization of the pathogenetic mechanisms of ovarian cancer and identification of the associated molecular regulators is imperative to improve the treatment of ovarian cancer.
GTPase-activating proteins (GAPs) are regulators of GTPase proteins and other domains, which directly or indirectly contribute to many cell functions and processes, including programmed cell death (PCD) [4]. Altered expression level of GAPs is associated with many types of cancer. For instance, ARHGAP5 and ARHGAP11A were found to be upregulated in metastatic colorectal cancers and gastrointestinal cancers, respectively [5, 6]. Of direct relevance to our work, Luo et al. [7] reported downregulation of a GAP named ARHGAP10 in ovarian cancer, while its overexpression was found to inhibit cancer cell adhesion, migration, invasion, and viability. Although their study showed that the anticancer effects of ARHGAP10 were mediated via inhibition of Cdc42 and activation of cancer cell apoptosis, the potential contribution of other signaling pathways and cell death mechanisms cannot be ruled out. Indeed, in the above-cited study, overexpression of ARHGAP10 was found to down-regulate PCNA, PARP1, and PARP2 [7], all of which are associated with another type of PCD–ferroptosis [8, 9, 10], which is also a newly emerging approach for cancer therapy [11].
Butyrate is a fatty acid that can regulate multiple cell cycle protein including GAPs [12]. Due to the inhibitory effect of butyrate on the proliferation of cancer cells (e.g., colorectal and gastric cancer [12, 13]), it was suggested as a promising medication supplement for cancer treatment. Although the most acknowledged mechanism of butyrate-induced tumor inhibition is via the mitochondrial apoptosis pathway [14], we noticed that butyrate can also trigger ferroptosis in certain types of cells [15].
The above body of evidence prompted us to study whether the cancer-inhibitory effects of ARHGAP10 and butyrate are in part due to ferroptosis. We found that ARHGAP10 directly facilitated ferroptosis of ovarian cancer cells, which inhibited cancer cell viability and tumor growth in mice. Treatment of ovarian cancer cells with sodium butyrate (SB) greatly elevated the transcription level of ARHGAP10, which in turn facilitated ferroptosis. These results indicated a direct contribution of both ARHGAP10 and butyrate to the ferroptosis of ovarian cancer cells and revealed a previously undiscovered signaling pathway between these two molecules, with SB transcriptionally regulating ARHGAP10. Overall, our study unraveled the ferroptosis induced by SB and ARHGAP10, which expands our understanding of cancer pathology and may inspire novel anticancer therapeutic strategies.
OVCAR3 and A2780 human ovarian cancer cell lines were acquired from the Chinese
Type Culture Collection, Chinese Academy of Sciences. The cells were cultured at
37 °C and 5% CO
The sequence of shRNA targeting ARHGAP10 (shARHGAP10) was GGTTCACAATTATCAGAAA. The shRNA was included in pLKO.1 lentiviral vector (Addgene, Watertown, MA, USA). Lentiviral constructs of pLKO.1-scramble shRNA (shNC), pLKO.1-shARHGAP10, pLVX-Puro empty vector (Clontech, Palo Alto, CA, USA), or pLVX-Puro-ARHGAP10 were co-transfected with viral packaging plasmids (psPAX2 and pMD2.G) into 293T cells. The viral supernatant was harvested after 48 h and filtered. A2780 cells were infected with pLVX-Puro-ARHGAP10 lentivirus while OVCAR3 cells were infected with pLKO.1-shARHGAP10 lentivirus with 8 µg/mL polybrene. Stable pools were obtained with 0.5 µg/mL puromycin (Sigma, St. Louis, MO, USA).
A2780 cells infected with pLVX-Puro-ARHGAP10 lentivirus were treated with or without 25 µM apoptosis inhibitor zVAD-fmk, 20 µM necroptosis inhibitor necrostatin-1 (Nec-1), or 5 µM ferroptosis inhibitor ferrostatin-1 (Fer; all from Selleck, Shanghai, China). Another set of A2780 cells was infected with pLVX-Puro-ARHGAP10 lentivirus and/or treated with 5 µM Fer-1.
OVCAR3 cells were infected with pLKO.1-shARHGAP10 lentivirus and/or treated with
1 µg/mL RSL3 (Selleck, Shanghai, China). Another set of OVCAR3 cells was
treated with 0.5 mM sodium butyrate with or without pLKO.1-shARHGAP10 lentivirus
infection. Cell Counting Kit-8 (CCK-8) Assay Kit (Dojindo Lab, Kumamoto, Japan)
was then used to measure cell viability in 96-well plates (2
A2780 cells infected with pLVX-Puro-ARHGAP10 lentivirus were treated with or without 25 µM zVAD-fmk, 20 µM Nec-1, or 5 µM Fer-1. Cell death rate was determined using the LDH cytotoxicity assay kit (Beyotime Biotechnology, Shanghai, China; C10016), as per the manufacturer’s instructions, and calculated as previously described [16].
Cells (3
Total RNA was extracted from cells using Trizol reagent (Invitrogen, Carlsbad,
CA, USA). The mRNA levels of ARHGAP10 were determined by quantitative RT-PCR
using GAPDH as a control. The primer sequences used were as follows: ARHGAP10,
5
Cells were lysed in ice-cold radioimmunoprecipitation assay buffer (50 mM
Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, 0.5% Na-deoxycholate)
containing protease inhibitors. Protein concentration was measured using a BCA
protein assay kit (Thermo Fisher Scientific). Equal amounts of cell lysates were
separated on sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels, transferred to polyvinylidene fluoride (PVDF) membranes, and analyzed by
Western blotting using an enhanced chemiluminescence system (Bio-Rad, Richmond,
CA, USA). The antibodies used are listed in Table 1. Band intensities were
analyzed using Image J (NIH, Bethesda, MD, USA) and normalized to
| Antibody name | Source | Cat. no | Dilution |
| ARHGAP10 | Proteintech Group | 55139-1-AP | 1:1000 |
| GPX4 | Abcam | ab125066 | 1:1000 |
| PTGS2 | Abcam | ab179800 | 1:1000 |
| H3K9ac | Abcam | ab32129 | 1:500 |
| Abcam | ab8226 | 1:1000 | |
| horseradish peroxidase-conjugated goat anti- rabbit IgG | ZSGB-BIO | ZB-2301 | 1:5000 |
| horseradish peroxidase-conjugated goat anti-mouse IgG | ZSGB-BIO | ZB-2305 | 1:5000 |
OVCAR3 cells were treated with 0.5 mM sodium butyrate or left untreated for 48
h, and then fixed with 1% formaldehyde. The fixed cells were sonicated for
chromatin fragmentation. The chromatin solutions were then incubated overnight
with anti-H3K9ac (Cell Signaling Technology, Danvers, MA, USA; 9649) or control
IgG antibody (Cell Signaling Technology; 3900) at 4 °C. H3K9ac binding
in the ARHGAP10 promoter region was quantified by PCR using the following
ARHGAP10 promoter primers: 5
Athymic Balb/c nude mice (age: 5 weeks) were acquired from the Slac Laboratory
Animal Co. Ltd. (Shanghai, China) and housed in a pathogen-free animal facility.
Mice were randomly assigned to control or experimental groups (6 per group).
A2780 cells infected with pLVX-Puro-ARHGAP10 lentivirus were injected
intraperitoneally into the flank of each mouse (2
After fixation and permeabilization, the tissues collected from the xenograft tumor were incubated with 1% bovine serum albumin for 30 min for blocking and then with anti-Ki67 (Abcam; ab243878) and Alexa Fluor 488-labeled Goat Anti-Mouse IgG (H + L) (Beyotime Biotechnology; A0423). DAPI (Beyotime Biotechnology; C1002) labeling was used for nuclear staining. Then, positively stained cells were visualized using a confocal laser scanning microscope (Leica Microsystems, Inc., Wetzlar, Germany).
All experiments were conducted in triplicate. Data were expressed as mean
To understand the effects of ARHGAP10 on ovarian cancer, we first used pLVX-Puro-ARHGAP10 lentivirus to induce the overexpression of ARHGAP10. A2780 cell line was used as an in vitro cell model of human ovarian cancer. After infection with pLVX-Puro-ARHGAP10 lentivirus, the A2780 cells showed much higher mRNA and protein expression of ARHGAP10 compared with non-infected cells (Control) or cell infected with empty lentivirus (Vector) (Fig. 1A,B), confirming the success of the infection.
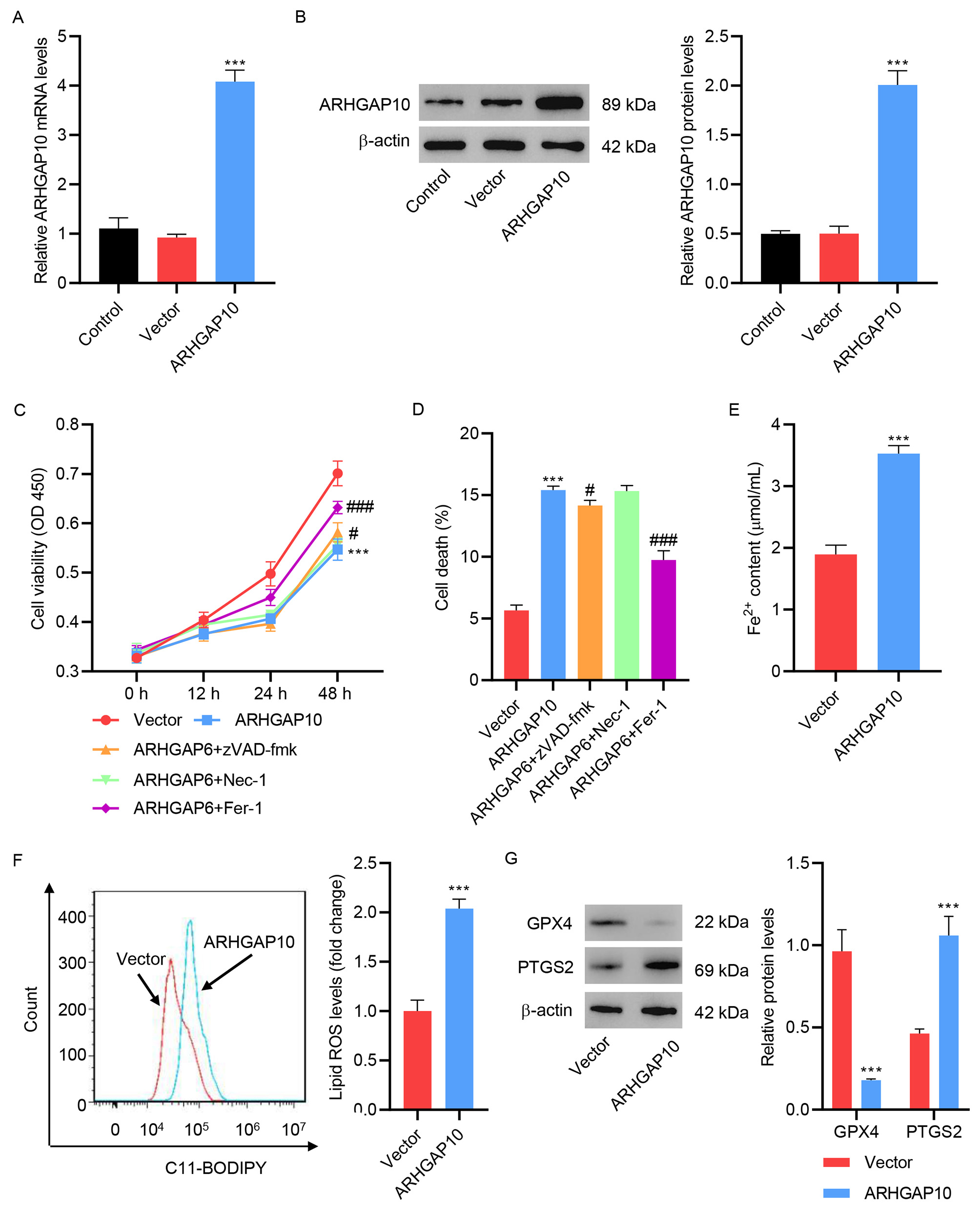 Fig. 1.
Fig. 1.ARHGAP10 overexpression promotes A2780 cell ferroptosis. (A)
mRNA and (B) protein levels of ARHGAP10 in A2780 cells infected with
pLVX-Puro-ARHGAP10 lentivirus or control lentivirus (n = 3). A2780 cells were
infected with pLVX-Puro-ARHGAP10 lentivirus and treated with 25 µM
apoptosis inhibitor zVAD-fmk, 20 µM necroptosis inhibitor Nec-1, or 10
µM ferroptosis inhibitor Fer-1, and the (C) cell viability and (D)
cell death were determined (n = 3). The (E) Fe
Consistent with a previous study [7], our results showed that ARHGAP10
overexpression caused a significant decrease in cell viability (Fig. 1C) and an
increase in cell death (Fig. 1D). To determine the cause of ARHGAP10
overexpression-induced cell death, several cell death inhibitors were tested.
Ferroptosis inhibitor Fer-1 markedly inhibited the ARHGAP10
overexpression-induced cell viability decrease and cell death. However, apoptosis
inhibitor zVAD-fmk only slightly inhibited ARHGAP6 overexpression-induced cell
viability decrease and cell death (Fig. 1C,D). To further examine the effects of
ferroptosis on ARHGAP10 overexpression-mediated cell death, Fe
To further confirm the triggering of ferroptosis by ARHGAP10 overexpression, we
tested the effect of Fer-1. Both in the absence and presence of ARHGAP10
overexpression, treatment with Fer-1 significantly increased cell viability (Fig. 2A), and decreased the Fe
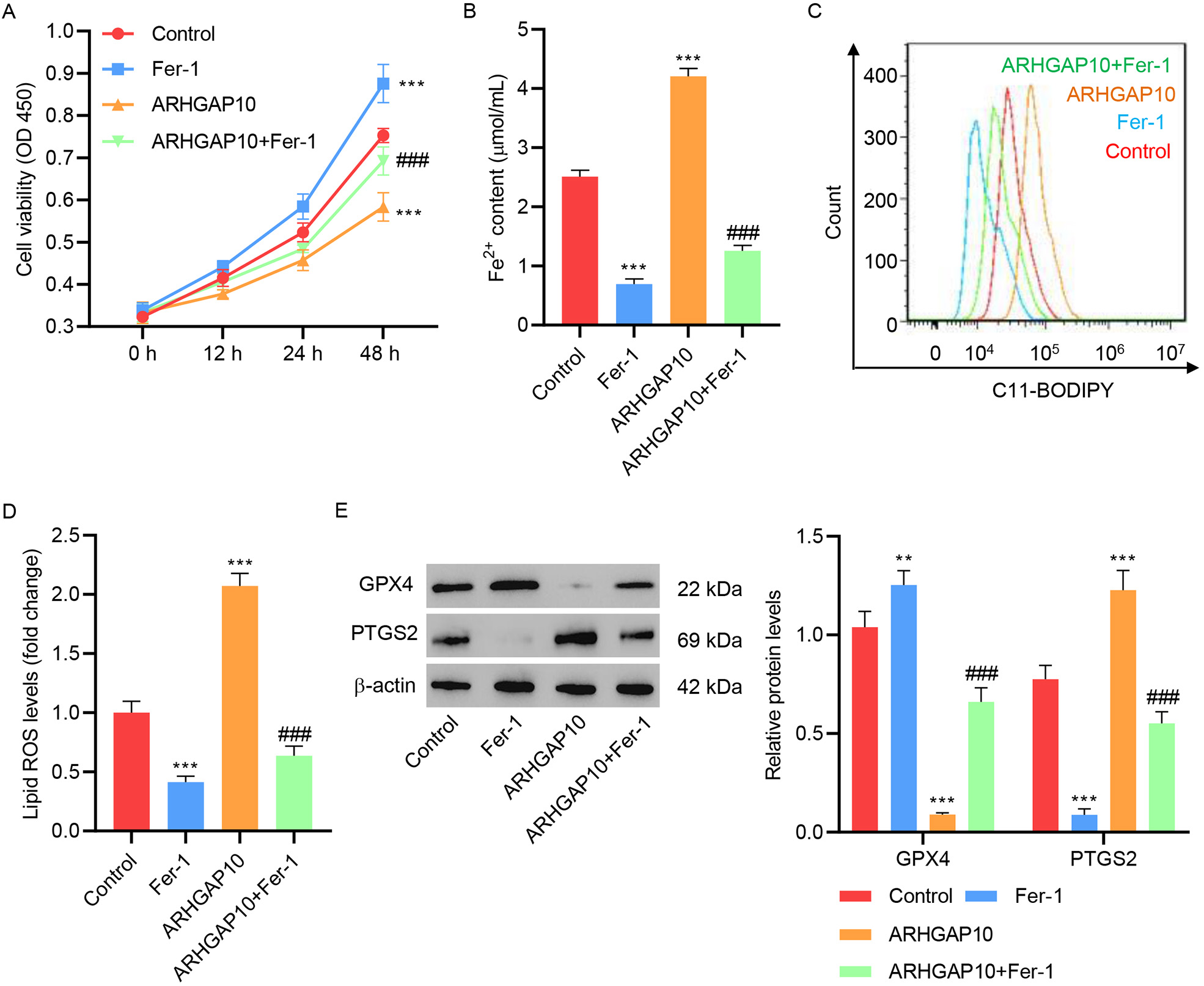 Fig. 2.
Fig. 2.Fer-1 inhibits ARHGAP10 overexpression-induced ferroptosis in
A2780 cells. (A) Cell viability, (B) Fe
In a previous study, ARHGAP10 was found to suppress tumor growth [7], which was argued to be contributed by apoptosis. However, other molecular mechanisms may also be involved in contributing to this effect. Based on our above results which showed that ARHGAP10 triggers ferroptosis in cancer cells, we hypothesized that it also contributes to tumor suppression.
To test our hypothesis, mice were injected with A2780 cells with or without ARHGAP10 overexpression to induce tumor development. Some of the mice in both groups were then treated with Fer-1 for 35 days, while the rest were left untreated. The development of tumors in each mouse was monitored for 35 days. Compared to the control group, tumors in mice treated with Fer-1 were larger and heavier, suggesting that ferroptosis is indeed a regulator of ovarian tumor growth (Fig. 3A–C). Notably, whereas ARHGAP10 overexpression significantly decreased tumor size and weight, these effects were reversed by Fer-1 (Fig. 3A–C).
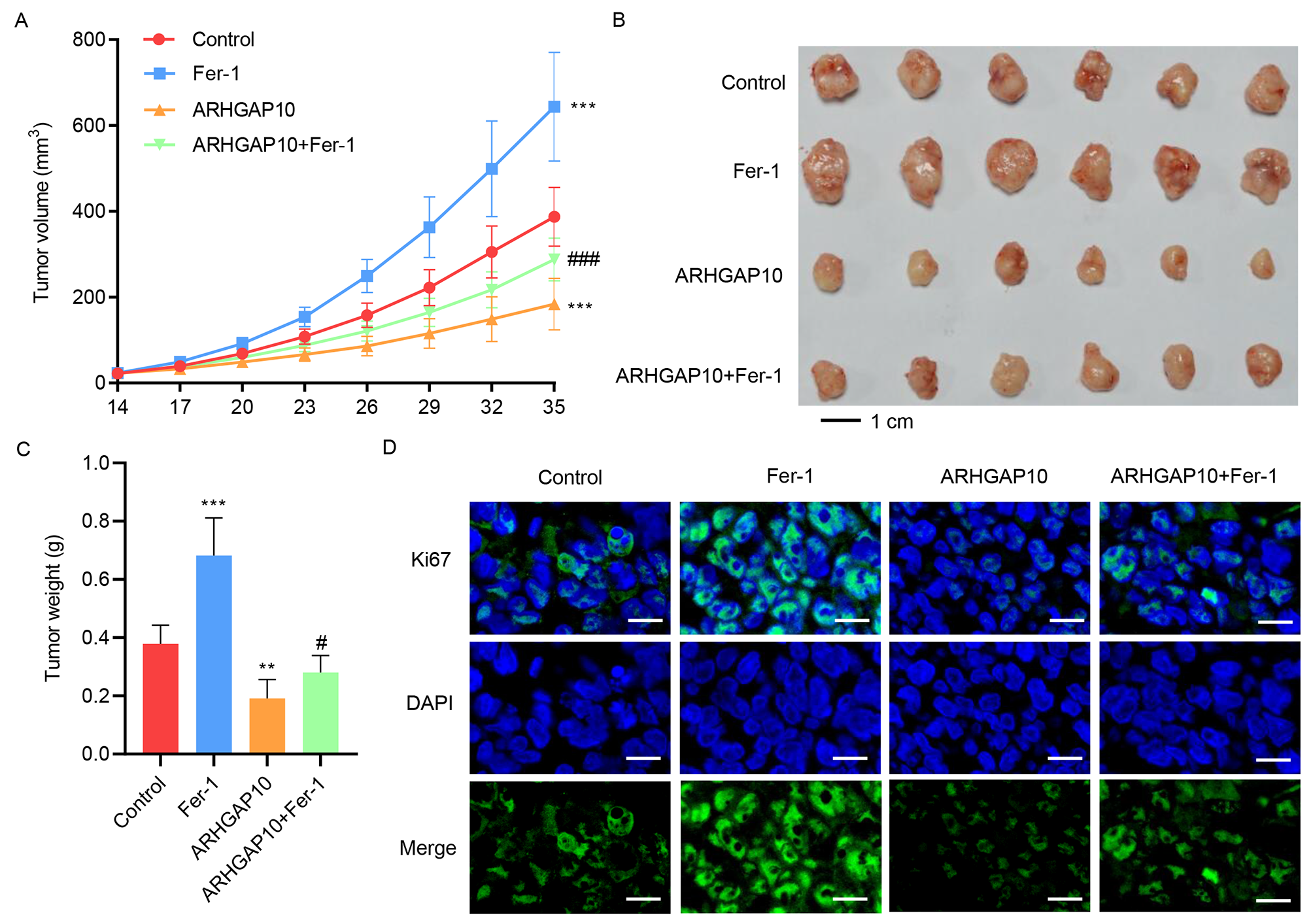 Fig. 3.
Fig. 3.Fer-1 inhibits ARHGAP10 overexpression-induced suppression of
tumor growth in vivo. Mice injected with A2780 cells were treated with
Fer-1 for 35 days. Tumor (A) volume, (B,C) weight, and (D) Ki67
immunofluorescence staining (scale bar, 20 µm) were quantified (n
= 6). **p
We also looked at immunofluorescence staining of Ki67, a commonly used biomarker of cell proliferation [20], in the xenograft tumor tissue. While Fer-1 greatly increased the signal intensity of Ki67 in the tumor cells, suggesting higher viability and greater proliferation, the signal intensity was drastically decreased by ARHGAP10 overexpression, and was restored to a level comparable to the control group by the copresence of ARHGAP10 overexpression and Fer-1 (Fig. 3D). These results indicated that ARHGAP10-induced suppression of tumor growth was in part via the ferroptosis pathway, which can be inhibited by Fer-1.
ARHGAP10 silencing is another way to re-confirm the observed relation between
ARHGAP10 and ferroptosis. A shRNA targeting ARHGAP10 (shARHGAP10) was developed
for this purpose and used to construct the pLKO.1-shARHGAP10 lentivirus.
Infection of OVCAR3 cells with the control lentivirus (shNC) had no effect on the
mRNA and protein expression levels of ARHGAP10. However, OVCAR3 cells infected
with pLKO.1-shARHGAP10 manifested much lower mRNA and protein expression levels
of ARHGAP10, confirming the specificity of pLKO.1-shARHGAP10 in ARHGAP10
silencing (Fig. 4A,B). Silencing of ARHGAP10 significantly increased cell
viability (Fig. 4C), Fe
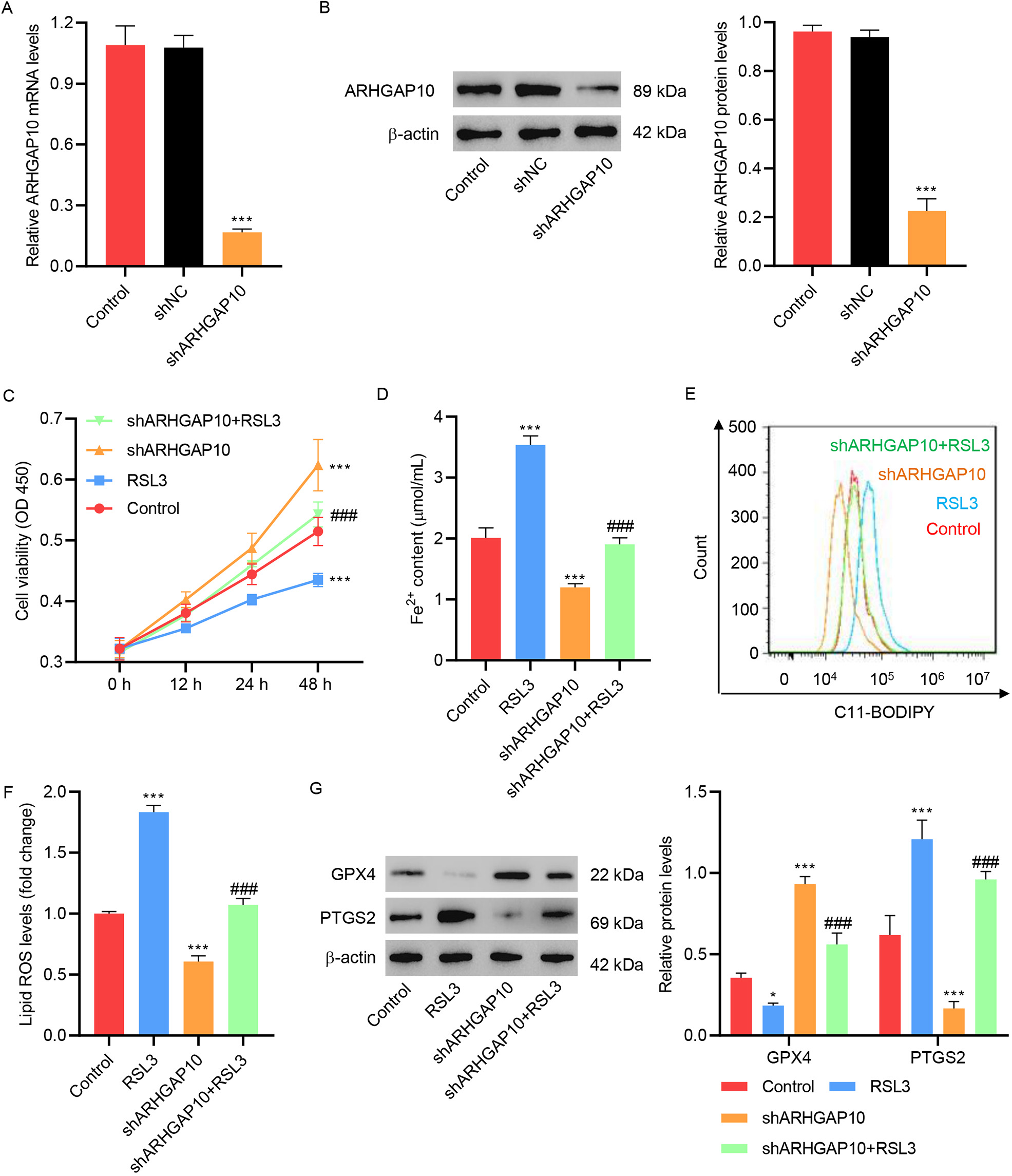 Fig. 4.
Fig. 4.RSL3 inhibits ARHGAP10 silencing-induced suppression of
ferroptosis in OVCAR3 cells. (A) mRNA and (B) protein levels of ARHGAP10 in
OVCAR3 cells infected with pLKO.1-shARHGAP10 lentivirus or control lentivirus (n
= 3). (C) Cell viability, (D) Fe
RSL3 is a facilitator of cell ferroptosis, which has been shown to inactivate GPX4 and stimulate ROS production in cancer cells [21, 22]. Here, we showed that RSL3 decreased the viability of OVCAR3 cells, increased the lipid ROS level, and decreased and increased the expression of GPX4 and PTGS2, respectively (Fig. 4C–G). Treatment of OVCAR3 cells with RSL3 reversed the effect of ARHGAP10 silencing so that the cell viability level, lipid ROS level as well as GPX4 and PTGS2 expression levels were at an intermediate level between the ARHGAP10-silenced group and RSL3-treated group (Fig. 4C–G).
Lastly, we investigated the potential relation of SB with ARHGAP10 in ferroptosis. Treatment of ovarian cancer cells with SB greatly increased the mRNA and protein expression levels of ARHGAP10 (Fig. 5A,B). SB also increased the expression level of H3K9ac, a marker of transcriptional activation of genes (Fig. 5B). This suggested that SB may enhance the expression of ARHGAP10 by regulating the transcription of its encoding gene. The results of the ChIP assay showed that SB can increase the binding of H3K9ac to the promoter region of ARHGAP10 (Fig. 5C,D).
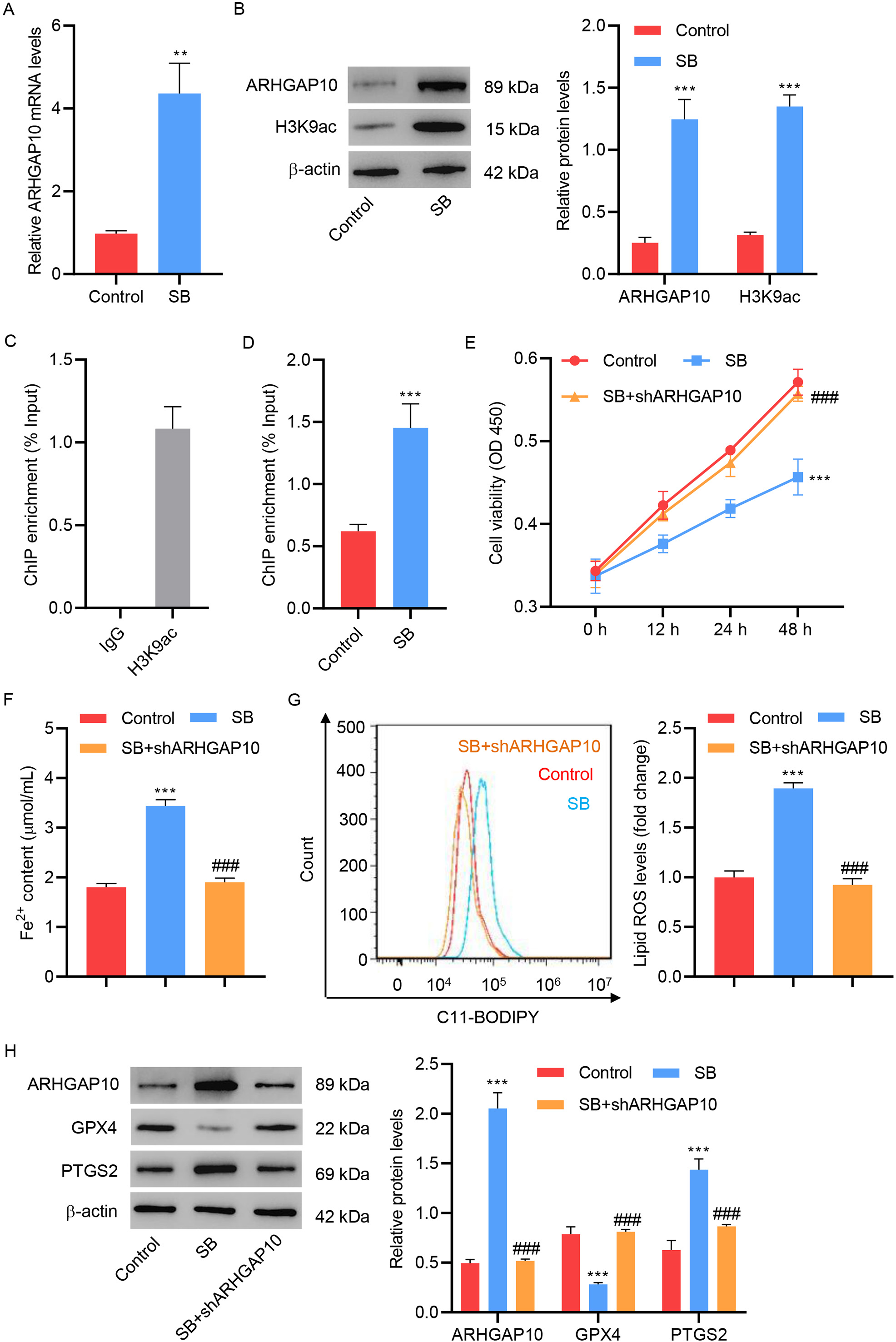 Fig. 5.
Fig. 5.Sodium butyrate promotes OVCAR3 cell ferroptosis by transcriptional regulation of ARHGAP10. (A) mRNA
levels of ARHGAP10 and (B) protein levels of ARHGAP10 and H3K9ac in OVCAR3 cells
treated with sodium butyrate (SB) (n = 3). (C) Chromatin from OVCAR3 cells was
immunoprecipitated using control IgG or H3K9ac antibody, and the results were
measured by quantitative PCR (n = 3). (D) Chromatin from OVCAR3 cells with or
without SB treatment were immunoprecipitated using the H3K9ac antibody, and the
results were measured by quantitative PCR (n = 3). (E) Cell viability, (F)
Fe
Consistent with the above finding, we further found that SB significantly
decreased the viability of OVCAR3 cells (Fig. 5E), increased the Fe
A key bottleneck in current cancer treatment, including conventional approaches like chemotherapy, or relatively novel ones like immunotherapy, is the development of resistance against these therapies [23]. Previous studies have found that cancer cells can undergo ferroptosis, a new type of PCD distinct from apoptosis, necrosis, or autophagy [24], in response to radiotherapy, chemotherapy, and immunotherapy [23], suggesting that activating cell ferroptosis may serve as a promising strategy to increase the efficacy of anticancer treatment. Of relevance to this work, ferroptosis has been proposed as an advanced strategy for treating ovarian cancer. For instance, ferroptosis inducers were found to increase the sensitivity of BRCA-proficient ovarian cancer cells and xenografts to pharmacologic PARP inhibitors [10]. In the present study, ARHGAP10 was found to facilitate ovarian cancer cell ferroptosis, which is transcriptionally regulated by SB. Altogether, these results highlight ARHGAP10 as a potential target for drug development against ovarian cancer and delineate the underlying mechanism by which SB induces ferroptosis.
Since its discovery in 2012 [25], ferroptosis is known to be mediated by at
least eight major pathways: the glutathione-GPX4 pathway, NADPH-FSP1-CoQ10
pathway, P53-mediated lipoxygenase and iPLA2
Interestingly, ARHGAP10 was only reported to enhance apoptosis in ovarian cancer cells, which was believed to be the sole mechanism of its induced tumor suppression [7]. Our work provides a paradigm shift by discovering a second mechanism that is based on ferroptosis. This suggests that targeting ARHGAP10 can have at least bifold effects for treating ovarian cancer because it can concurrently facilitate both apoptosis and ferroptosis. Furthermore, our discovery opens several future directions of research. First, it is intriguing to ask whether ARHGAP10 is also associated with other types of PCD. In addition, its relationship with necrosis has not been discussed before. Figuring out the exact contributions of ARHGAP10 to PCD will enable an in-depth understanding of its mechanisms of action and facilitate its targeting for therapeutic purposes. Second, different types of PCD can have synergistic effects on each other to achieve stronger effects. For example, autophagy can facilitate ferroptosis by degrading ferritin [28]. We therefore, are curious whether the apoptosis and ferroptosis signaling pathways downstream of ARHGAP10 have any overlapping or mutual effects. Lastly, recent studies have shown that ARHGAP10 is associated with various cancers, such as head and neck squamous cell carcinoma [29], gliomas [30], and prostate cancer [31]. It would be extremely meaningful to expand our study and explore whether ARHGAP10 can also facilitate ferroptosis in other types of cancer.
SB has shown great potential in cancer treatment [12, 13, 32]. Similar to ARHGAP10, the tumor inhibitory effect of SB is also believed to be due to its proapoptotic effects. Specifically, by increasing the expression of DAPK, SB potentially increases the expression of several cell-cycle inhibitors (e.g., CDKN1A, CDKN1B) and the activity of several proapoptotic genes (e.g., BAX, BAK, and BIK) [13]; it also down-regulates certain cancer-facilitating molecules (e.g., Thioredoxin-1) [33]. Here we show that ferroptosis may also contribute to SB-induced tumor suppression, which emphasizes the therapeutic potential of SB. Furthermore, by pinpointing ARHGAP10 as a necessary signaling molecule mediating the effect of SB on ferroptosis, our findings seem to suggest a pivotal role of ARHGAP10 in the signaling network of ferroptosis, which would require future investigations.
In this study, ARHGAP10 was found to promote ferroptosis of ovarian cancer cells via a GPX4-dependent pathway to suppress tumor development. Furthermore, SB was found to regulate the transcription of ARHGAP10, thereby also contributing to ovarian cancer cell ferroptosis. To the best of our knowledge, this is the first study to report ferroptosis-related functions of SB and ARHGAP10. Our study expands our current understanding of the antitumor effects of SB and ARHGAP10 and emphasizes the SB/ARHGAP10/GPX4 signaling pathway as a promising therapeutic target in the context of ovarian cancer.
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Conceptualization: HK and JS; Methodology: JS, JH, XS, HH, LC and ZZ; Formal analysis and investigation: JH, XS, YS and XZ; Writing-original draft preparation: HK; Writing-review and editing: LC and YS; Funding acquisition: YS; Supervision: XZ, LC and YS. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All experimental procedures were performed in accordance with the Guidelines for the Animal Experimentation Ethics Committee of Shanghai Pudong Hospital, Fudan University Pudong Medical Center [(2021)No.(QKW-02)].
Not applicable.
This study was funded by the Science and Technology Development Fund of Shanghai Pudong (PKJ2021-Y36) and Key Specialty Construction Project of Pudong Health and Family (PWZzk2022-21) and supported by the Project of Key Medical Specialty and Treatment Center of Pudong Hospital of Fudan University (Tszb2023-03).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.